Blog
2017.12.18
Release Chainer Chemistry: A library for Deep Learning in Biology and Chemistry
Tag
Kosuke Nakago
Engineer
* Japanese blog is also written here.
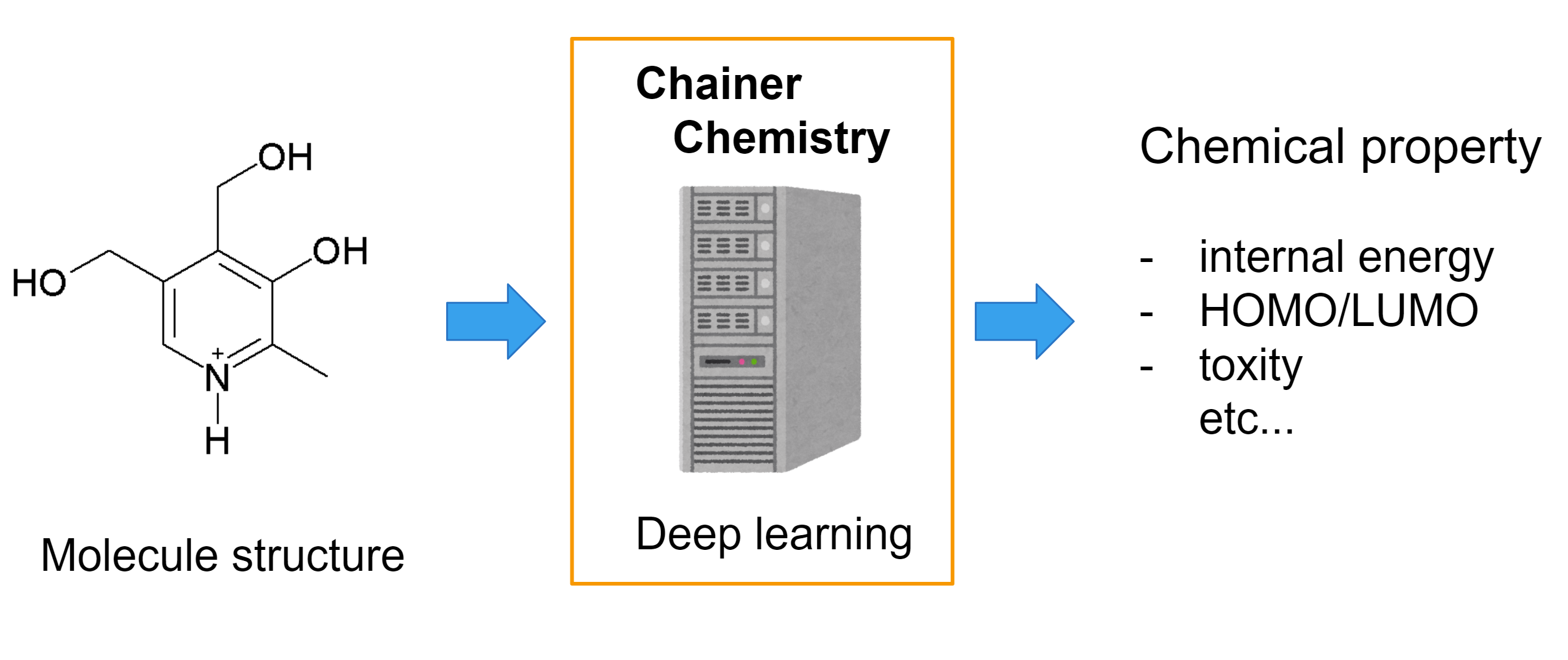
We released Chainer Chemistry, a Chainer [1] extension to train and run neural networks for tasks in biology and chemistry.
- Github page: https://github.com/pfnet-research/chainer-chemistry
- Documentation: https://chainer-chemistry.readthedocs.io
The library helps you to easily apply deep learning on molecular structures.
For example, you can apply machine learning on toxicity classification tasks or HOMO (highest occupied molecular orbital) level regression task with compound input.
The library was developed during the PFN 2017 summer internship, and part of the library has been implemented by an internship student, Hirotaka Akita at Kyoto University.
Supported features
Graph Convolutional Neural Network implementation
The recently proposed Graph Convolutional Network (Refer below for detail) opened the door to apply deep learning on “graph structure” input, and the Graph Convolution Networks are currently an active area of research. We implemented several Graph Convolution Network architectures, including the network introduced in this year’s paper.
The following models are implemented:
- NFP: Neural Fingerprint [2, 3]
- GGNN: Gated-Graph Neural Network [4, 3]
- WeaveNet: Molecular Graph Convolutions [5, 3]
- SchNet: A continuous-filter convolutional Neural Network [6]
Common data preprocessing/research dataset support
Various datasets can be used with a common interface with this library. Also, some research datasets can be downloaded automatically and preprocessed.
The following datasets are supported:
- QM9 [7, 8]: dataset of organic molecular structures with up to nine C/O/N/F atoms and their computed physical property values. The values include HOMO/LUMO level and internal energy. The computation is B3LYP/6-31G level of quantum chemistry.
- Tox21 [9]: dataset of toxicity measurements on 12 biological targets
Train/inference example code is available
We provide example code for training models and inference. You can easily try training/inference of the models implemented in this library for quick start.
Background
In the new material discovery/drug discovery field, simulation of molecule behavior is important. When we need to take quantum effects into account with high precision, DFT (density functional theory) is widely used. However it requires a lot of computational resources especially for big molecules. It is difficult to apply simulation on many molecule structures.
There is a different approach from the machine learning field: learn the data measured/calculated in previous experiments, and predict the unexperimented molecule’s chemical property. The neural network may calculate the prediction faster than the quantum simulation.
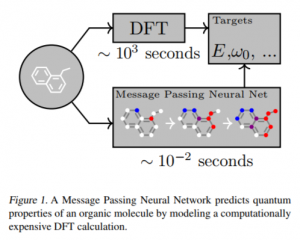
Cited from “Neural Message Passing for Quantum Chemistry”, Justin et al. https://arxiv.org/pdf/1704.01212.pdf
An important question is how to deal with the input/output of compounds in order to apply deep learning. The main problem is that all molecular structures have variable numbers of atoms, represented as different graph structures, while conventional deep learning methods deal with a fixed size/structured input.
However “Graph Convolutional Neural Network” is proposed to deal with graph structure for input.
What is a Graph Convolutional Neural Network
Convolutional Neural Networks introduce “convolutional” layers which applies a kernel on local information in an image. It shows promising results on many image tasks, including classification, detection, segmentation, and even image generation tasks.
Graph Convolutional Neural Networks introduce a “graph convolution” operation which applies a kernel among the neighboring nodes on the graph, to deal with graph structure.
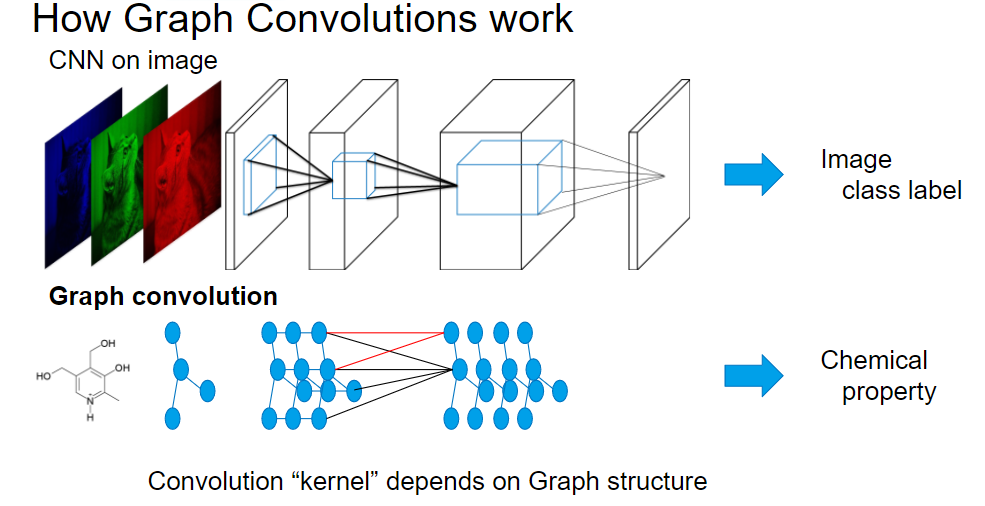
CNN deals with an image as input, whereas Graph CNN can deal with a graph structure (molecule structure etc) as input.
Its application is not limited to molecule structure. “Graph structures” can appear in many other fields, including social networks, transportation etc, and the research of graph convolutional neural network applications is an interesting topic. For example, [10] applied graph convolution on image, [11] applied it on knowledge base, [12] applied it on traffic forecasting.
Target users
- Deep learning researchers
This library provides latest Graph Convolutional Neural Network implementation
Graph Convolution application is not limited to Biology & Chemistry, but various kinds of fields. We would like many people to use this library. - Material/drug discovery researchers
The library enables the user to build their own model to predict various kinds of chemical properties of a molecule.
Future plan
This library is still a beta version, and in active development. We would like to support the following features:
- Provide pre-trained models for inference
- Add more datasets
- Implement more networks
We prepared a Tutorial to get started with this library, please try and let us know if you have any feedback.
Reference
[1] Tokui, S., Oono, K., Hido, S., & Clayton, J. (2015). Chainer: a next-generation open source framework for deep learning. In Proceedings of workshop on machine learning systems (LearningSys) in the twenty-ninth annual conference on neural information processing systems (NIPS) (Vol. 5).
[2] Duvenaud, D. K., Maclaurin, D., Iparraguirre, J., Bombarell, R., Hirzel, T., Aspuru-Guzik, A., & Adams, R. P. (2015). Convolutional networks on graphs for learning molecular fingerprints. In Advances in neural information processing systems (pp. 2224-2232).
[3] Gilmer, J., Schoenholz, S. S., Riley, P. F., Vinyals, O., & Dahl, G. E. (2017). Neural message passing for quantum chemistry. arXiv preprint arXiv:1704.01212.
[4] Li, Y., Tarlow, D., Brockschmidt, M., & Zemel, R. (2015). Gated graph sequence neural networks. arXiv preprint arXiv:1511.05493.
[5] Kearnes, S., McCloskey, K., Berndl, M., Pande, V., & Riley, P. (2016). Molecular graph convolutions: moving beyond fingerprints. Journal of computer-aided molecular design, 30(8), 595-608.
[6] Kristof T. Schütt, Pieter-Jan Kindermans, Huziel E. Sauceda, Stefan Chmiela, Alexandre Tkatchenko, Klaus-Robert Müller (2017). SchNet: A continuous-filter convolutional neural network for modeling quantum interactions. arXiv preprint arXiv:1706.08566
[7] L. Ruddigkeit, R. van Deursen, L. C. Blum, J.-L. Reymond, Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17, J. Chem. Inf. Model. 52, 2864–2875, 2012.
[8] R. Ramakrishnan, P. O. Dral, M. Rupp, O. A. von Lilienfeld, Quantum chemistry structures and properties of 134 kilo molecules, Scientific Data 1, 140022, 2014.
[9] Huang R, Xia M, Nguyen D-T, Zhao T, Sakamuru S, Zhao J, Shahane SA, Rossoshek A and Simeonov A (2016) Tox21 Challenge to Build Predictive Models of Nuclear Receptor and Stress Response Pathways as Mediated by Exposure to Environmental Chemicals and Drugs. Front. Environ. Sci. 3:85. doi: 10.3389/fenvs.2015.00085
[10] Michaël Defferrard, Xavier Bresson, Pierre Vandergheynst (2016), Convolutional Neural Networks on Graphs with Fast Localized Spectral Filtering, NIPS 2016.
[11] Michael Schlichtkrull, Thomas N. Kipf, Peter Bloem, Rianne van den Berg, Ivan Titov, Max Welling (2017) Modeling Relational Data with Graph Convolutional Networks. arXiv preprint arXiv: 1703.06103
[12] Yaguang Li, Rose Yu, Cyrus Shahabi, Yan Liu (2017) Diffusion Convolutional Recurrent Neural Network: Data-Driven Traffic Forecasting. arXiv preprint arXiv: 1707.01926
Tag